PATHOLOGIES
Bethlem MyopathyBETHLEM MYOPATHY
WHAT IS
Bethlem myopathy is an autosomal dominant myopathy classified as a congenital form of muscular dystrophy, it is caused by a mutation in one of the three genes coding for type VI collagen, described for the first time in 1966 by J. Bethlem.
In some rare cases it only appears in an autosomal recessive form.
DIAGNOSIS
The first step for a timely diagnosis of the Bethlem Myopathy is the observation of the aformentioned common clinical signs. The concentration of creatine kinase (CK) may be normal or mildly elevated.
A muscle biopsy with immunostaining of collagen type VI or skin biopsy.may be necessary.
It can also be confirmed by genetic testing, with the search for mutations in the genes involved.
CLINICAL SIGNS
The first symptoms of the Bethlem myopathy can appear at any time from birth through adulthood and they can be extremely variable. In childhood these symptoms can be muscle weakness, hypotonia, delayed motor milestones and contractures in the knees, ankles, hips and elbows.
Bethlem myopathy is an apparently rare early-onset benign autosomal dominant limb-girdle myopathy with contractures of the fingers.
Since it progresses very slowly, only part of affected require walking aids (cane, wheelchair, etc..) after 50-60 years.
Some patients showed, roughly past their 50s, of breathing problems making it necessary to use a BiPAP machine while sleeping.
There is also the possibility that lordosis or scoliosis, follicular hyperkeratosis. hypermobility of the joints, keloid formation on wounds may occur.

Bethlem-Mani-Hands-Col6.it-collagene-6-vi
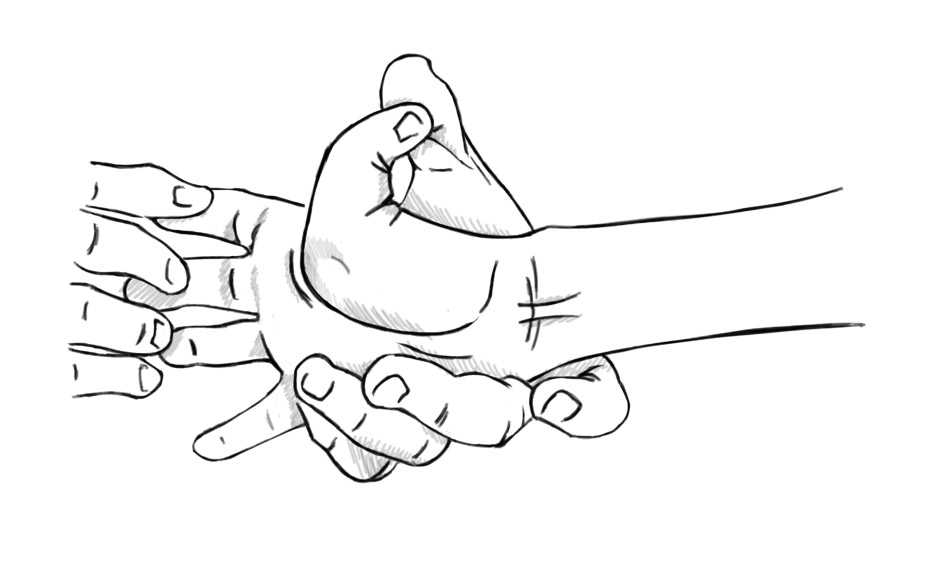
Joint hypermobility
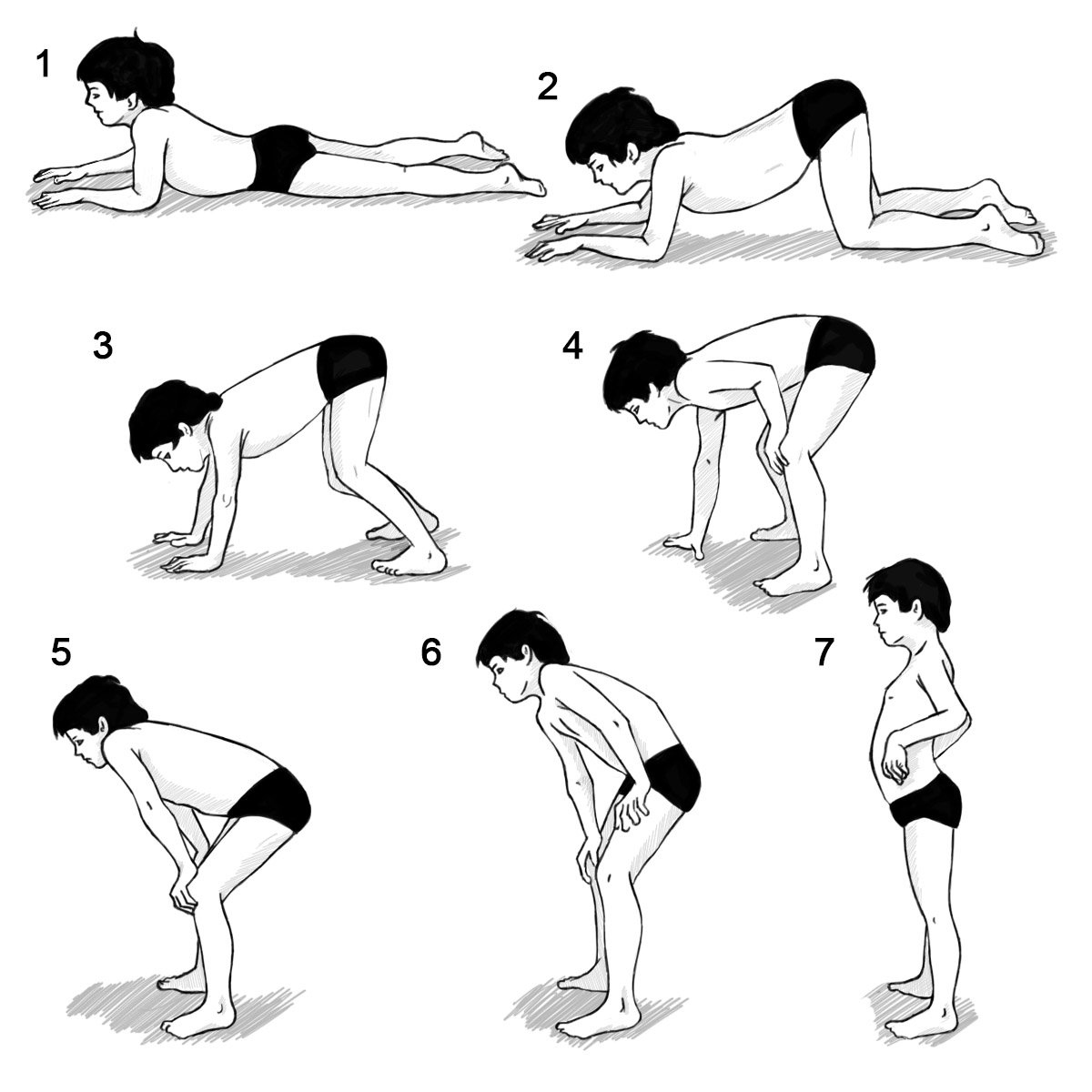
Gowers’ Sign
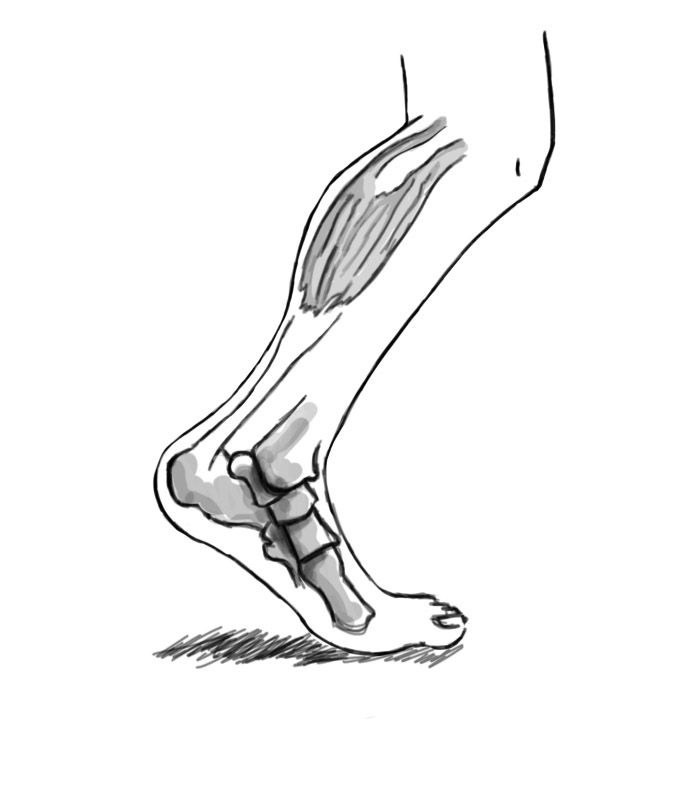
Retractions in the Achilles tendons
Inheritance
Bethlem myopathy is inherited in an autosomal dominant (one mutation is sufficient to cause the disease) or recessive (two mutations are required) manner; the myosclerotic variant is inherited in a recessive manner only. If the inheritance is dominant, it may happen that one of the parents is affected (the one who transmitted the mutation) or both are unaffected if the mutation arose de novo.
If the inheritance is recessive, the parents of the affected person are usually healthy carriers. The offspring of the affected person are healthy carriers.
Once the mutation has been identified, it is possible to assess the risk of transmission of the disease and propose a prenatal diagnosis in high-risk pregnancies.
Treatment
As of today, there aren’t drug treatments. Is possible to relieve the effects of the tendon contractures (only in some isolated cases) through elongating surgeries and/or physiotherapy.
Research
Some promising studies have been made in the last few years that have helped better understand the disease mechanism:
1. An immunosuppressant, generally used after transplantation (cyclosporine A) to prevent rejection, has granted promising results, demonstrating to act as an inhibitor of the mitochondrial permeability transition pore.
However, because of its immunosuppressive effect, this substance was not taken into consideration as a possible therapy for mitochondrial dysfunction. A drug was subsequently designed, analogue of cyclosporine, but without immunosuppressive power. (Aka Alisporivir Debio-025).
During the development of the drug, because of three pancreatitis cases and one patient dying during clinical trials for hepatitis C, the study was stopped to determine whether the drug could have been responsible for this side effect.
2. A research is in progress at this time, concerning the study of autophagy involvement that seems to be defective in muscular dystrophies patients affect by the collagen VI.