Fonte: https://www.mdpi.com/2218-273X/15/10/1426
Il collagene di tipo VI è un componente della matrice extracellulare codificato dai geni COL6A1, COL6A2 e COL6A3. Le varianti patogenetiche in questi geni sono associate a diverse miopatie da deficit di collagene VI, tra cui la distrofia muscolare congenita di Ullrich, la miopatia di Bethlem e le forme intermedie. Abbiamo analizzato il panorama delle mutazioni nei geni COL6A in 138 pazienti italiani affetti da una miopatia correlata al collagene VI. La coorte comprendeva 44 pazienti con distrofia di Ullrich, 9 con forma intermedia, 61 con miopatia di Bethlem e 21 con una forma intermedia tra Bethlem e intermedia; inoltre, 3 pazienti presentavano una miopatia con miosclerosi.
Sono state identificate 104 varianti diverse: 26 nel gene COL6A1, 52 nel gene COL6A2 e 26 nel gene COL6A3. Le varianti osservate includono mutazioni missenso, alterazioni dello splicing, piccole inserzioni o delezioni, mutazioni frameshift e mutazioni nonsense. Le sostituzioni dell’aminoacido glicina nel dominio a tripla elica della proteina collagene VI risultano essere le più frequenti e sono presenti in tutti i tipi di manifestazione clinica. L’analisi genetica ha messo in evidenza uno scenario mutazionale peculiare e un’associazione fenotipica specifica del gene COL6A2 rispetto a COL6A1 e COL6A3, probabilmente riconducibile a una diversa storia evolutiva. Studiare il panorama mutazionale di varianti che si manifestano in malattie ultrarare, come le miopatie da deficit di collagene VI, è fondamentale per comprendere meglio il profilo genetico di queste condizioni e approfondire la conoscenza della struttura e dell’evoluzione dei geni coinvolti.
1. Introduzione
Il collagene di tipo VI è un componente della matrice extracellulare codificato dai geni COL6A1, COL6A2 e COL6A3. I geni COL6A1 e COL6A2 si trovano sul cromosoma 21, mentre COL6A3 è localizzato sul cromosoma 2. Il gene COL6A1 è formato da 35 esoni distribuiti su una regione di circa 23 mila paia di basi e viene trascritto in un unico messaggero. Il gene COL6A2 (composto da 28 esoni su circa 35 mila paia di basi) e il gene COL6A3 (che comprende 44 esoni su circa 100 mila paia di basi) vengono trascritti in diverse varianti grazie a un ampio fenomeno di splicing alternativo, cioè un processo che permette la produzione di più proteine a partire dallo stesso gene. Il collagene di tipo VI è costituito da tre catene principali, chiamate alfa 1, alfa 2 e alfa 3, codificate rispettivamente dai geni COL6A1, COL6A2 e COL6A3.
Queste catene si organizzano in una rete di microfibrille che svolge un ruolo fondamentale nel collegare la membrana basale ai tessuti circostanti della matrice extracellulare.
Ogni catena contiene un dominio centrale a tripla elica, formato da una sequenza ripetuta di tre amminoacidi (glicina, un amminoacido qualsiasi e prolina o idrossiprolina), affiancato da ampie regioni terminali alle due estremità che presentano somiglianze con alcune parti del fattore di von Willebrand, una proteina coinvolta nella coagulazione del sangue.
Il collagene di tipo VI è presente in molti tessuti dell’organismo, con la maggiore concentrazione nei tessuti connettivi, come ossa, pelle, tendini, cartilagine e nei fibroblasti interstiziali dei muscoli scheletrici. È stato inoltre osservato nel sistema nervoso centrale e periferico, nell’intestino, nei polmoni, nel tessuto adiposo, nelle isole pancreatiche, nei follicoli ovarici, nei glomeruli renali, nei vasi sanguigni e nella cornea.
Le mutazioni dei geni COL6A1, COL6A2 e COL6A3 provocano malattie dei muscoli scheletrici conosciute come miopatie da deficit di collagene di tipo VI. Queste malattie rappresentano una delle forme più comuni di distrofia muscolare congenita, identificate in circa un quinto dei pazienti italiani con questa diagnosi. Le miopatie da collagene di tipo VI comprendono un ampio spettro di manifestazioni cliniche, che vanno dalla forma più grave, la distrofia muscolare congenita di Ullrich, alla forma più lieve, la miopatia di Bethlem. Esistono anche forme intermedie di diversa gravità, e di recente è stata proposta una nuova categoria, chiamata miopatia intermedia da collagene di tipo VI, per i pazienti con caratteristiche cliniche a metà tra le due principali.
Un’altra forma, caratterizzata soprattutto da contratture muscolari diffuse, è definita miopatia con miosclerosi.
Diversi studi condotti a livello nazionale hanno descritto le mutazioni genetiche che colpiscono i geni COL6A. Le sostituzioni dell’amminoacido glicina nel dominio a tripla elica della proteina rappresentano il tipo di mutazione più comune, circa il 30% di tutte le varianti patogenetiche note.
Sono state inoltre individuate altre mutazioni puntiformi, alterazioni che interrompono la sequenza del gene, modifiche del meccanismo di splicing e, più raramente, ampie delezioni nel DNA. È stata anche identificata una variante ricorrente all’interno del gene COL6A1 che causa l’inserimento anomalo di una sequenza aggiuntiva nel messaggero. È interessante notare che queste varianti non sono specifiche di una popolazione o di un gruppo etnico.
Nei geni COL6A1, COL6A2 e COL6A3 si riscontrano sia mutazioni dominanti (che causano la malattia anche se presenti in una sola copia del gene) sia mutazioni recessive (che richiedono la presenza di due copie alterate). La gravità della malattia dipende dal modo in cui la mutazione influisce sulla struttura complessa del collagene di tipo VI e sul suo processo di assemblaggio. Oltre alle forme ereditarie, più della metà dei casi di distrofia muscolare congenita di Ullrich è dovuta a mutazioni nuove, non trasmesse dai genitori. Queste mutazioni, spesso localizzate nella parte iniziale del dominio a tripla elica della proteina, hanno un effetto negativo dominante, poiché impediscono alla catena alterata di partecipare correttamente alla formazione del collagene. Al contrario, nelle forme recessive più rare, le mutazioni localizzate verso la parte finale della catena impediscono la corretta formazione delle unità di base della proteina, bloccandone l’assemblaggio.
Il presente studio condotto in Italia descrive in modo approfondito le mutazioni dei geni COL6A in un gruppo di 138 pazienti affetti da miopatia da deficit di collagene di tipo VI. La diagnosi genetica è fondamentale per comprendere il legame tra alterazione genetica e manifestazione clinica, favorire la scoperta di nuovi indicatori biologici e permettere ai pazienti di accedere a terapie personalizzate.
2. Materiali e metodi
Coorte di pazienti
Sono stati studiati 138 pazienti con diagnosi clinica e genetica confermata di miopatia da deficit di collagene di tipo VI, provenienti da 21 centri diagnostici italiani: 13 situati nel Nord Italia, 5 nel Centro e 3 nel Sud. Il periodo di raccolta dei dati è compreso tra il 2007 e il 2022, pari a 15 anni di osservazione. Lo studio è stato condotto nell’ambito della normale procedura diagnostica e con finalità di diagnosi genetica. In ciascun centro è stato ottenuto il consenso etico come parte della prassi di routine per la diagnosi dei geni COL6A, e la ricerca è stata approvata dal Comitato Etico di Area Vasta Emilia Centro (numero di protocollo 66/2020/Oss/AOUFe, del 23 gennaio 2020).
I familiari affetti dei pazienti principali (probandi) non sono stati inclusi nel presente studio.
Il quadro clinico dei pazienti è stato definito in base ai criteri stabiliti durante il 166º Workshop Internazionale ENMC sulle miopatie da collagene di tipo VI, secondo i quali:
- la distrofia muscolare congenita di Ullrich si riferisce a pazienti che non hanno mai camminato o hanno perso la capacità di camminare entro i 12 anni di età;
- la miopatia di Bethlem riguarda pazienti che mantengono la deambulazione in età adulta (oltre i 19 anni);
- la forma intermedia si riferisce a pazienti che perdono la capacità di camminare durante l’adolescenza (tra i 13 e i 19 anni) [20].
Il fenotipo di miopatia con miosclerosi è stato anch’esso considerato, così come un fenotipo intermedio/miopatia di Bethlem (definito per pazienti ancora in grado di camminare al momento dell’ultima valutazione clinica, effettuata tra i 12 e i 19 anni).
Analisi genetica
I test genetici sono stati eseguiti mediante sequenziamento tradizionale (Sanger) e/o sequenziamento di nuova generazione con pannelli personalizzati. In alcuni casi selezionati è stata utilizzata una microarray CGH personalizzata. L’analisi dei geni COL6A con sequenziamento di nuova generazione è stata effettuata utilizzando un pannello personalizzato chiamato “NEUROMIO”, che comprende 171 geni neuromuscolari: 80 per neuropatie ereditarie, 46 per miopatie congenite, 42 per distrofie muscolari e 3 per malattie dei motoneuroni.
La preparazione delle librerie è stata eseguita con il metodo Illumina DNA Prep con arricchimento del target, seguita da sequenziamento paired-end di 150 basi sul sistema MiSeqTMDx (Illumina Inc., San Diego, California, USA). La copertura media ottenuta era di circa 500 letture per base. La copertura complessiva del pannello era del 99,8% con profondità di lettura superiore a 20 letture per base; per i geni COL6A la copertura era del 100%. L’analisi della sequenza includeva gli esoni codificanti e le regioni introniche adiacenti (±50 basi a monte e a valle di ciascun esone). Sono state progettate sonde specifiche per garantire la corretta analisi dell’intron 11 del gene COL6A1.
L’analisi dei dati grezzi comprendeva allineamento delle sequenze, annotazione, filtraggio e selezione delle varianti. Le varianti identificate, come polimorfismi a singolo nucleotide, inserzioni/delezioni e variazioni del numero di copie, sono state annotate utilizzando il software Emedgene, basato sul genoma umano di riferimento GRCh37. Per la visualizzazione della qualità dei dati e della profondità di lettura è stato utilizzato anche il software Integrative Genomics Viewer. Le varianti con frequenza superiore allo 0,1% nei database di popolazione, come Exome Variant Server, Exome Aggregation Consortium e gnomAD, sono state considerate comuni e escluse dall’analisi.
Tutte le varianti identificate sono state classificate secondo le linee guida dell’American College of Medical Genetics and Genomics in cinque categorie: patogenetiche, probabilmente patogenetiche, di significato incerto, probabilmente benigne e benigne. Strumenti come Varsome, ClinVar e Franklin sono stati utilizzati per raccogliere informazioni aggiornate sulle varianti. Le varianti considerate patogenetiche o probabilmente patogenetiche sono state incluse nell’analisi, così come le varianti di significato incerto che soddisfacevano il criterio di essere assenti nei controlli secondo le linee guida ACMG. La conferma molecolare delle varianti è stata effettuata mediante sequenziamento Sanger standard su analizzatori automatici (Applied Biosystems 3130xl e/o 3500DX, Thermo Fisher Scientific Inc., Waltham, Massachusetts, USA).
3. Risultati
I dati clinici e genetici dei pazienti con varianti nei geni COL6A1, COL6A2 e COL6A3 sono riportati nella Tabella 1. Un riepilogo delle caratteristiche delle varianti di significato incerto identificate nei geni COL6A1, COL6A2 e COL6A3 è riportato nella Tabella S1 (Fonte: https://www.mdpi.com/2218-273X/15/10/1426).
3.1 Varianti identificate nei geni COL6A
Sono state identificate 26 varianti diverse del gene COL6A1 in 48 pazienti (pari al 35% della coorte totale): 45 eterozigoti, 1 eterozigote composto e 2 omozigoti, con predominanza di varianti di splicing e missenso. Nel gene COL6A2 sono state rilevate 52 varianti diverse in 56 pazienti (40% della coorte totale): 32 eterozigoti, 12 eterozigoti composti e 12 omozigoti, comprendendo tutti i tipi di mutazioni, con predominanza di varianti missenso. Infine, nel gene COL6A3 sono state identificate 26 varianti, principalmente di tipo missenso, in 34 pazienti (25% della coorte): 31 eterozigoti, 1 eterozigote composto e 2 omozigoti (Figura 1a,b – Fonte: https://www.mdpi.com/2218-273X/15/10/1426).
Figura 1 – Fonte: https://www.mdpi.com/2218-273X/15/10/1426
a) Distribuzione dei geni COL6A tra i pazienti della nostra coorte. Per ciascun gene, è riportata la proporzione di pazienti eterozigoti, eterozigoti composti e omozigoti.
b) Tipologia e numero delle varianti nei tre geni COL6A.
Per tutte le varianti identificate è stata effettuata un’analisi di predizione della patogenicità secondo i criteri dell’American College of Medical Genetics and Genomics, con la seguente classificazione:
- 41 varianti patogenetiche
- 39 varianti probabilmente patogenetiche
- 24 varianti di significato incerto, che soddisfano il criterio PM2 delle linee guida (Tabella 1 e Tabella S1)
3.2 Geni COL6A e fenotipi
Nel complesso, nella nostra coorte sono stati identificati:
- 44 pazienti (32%) con distrofia muscolare congenita di Ullrich
- 9 pazienti (7%) con forma intermedia
- 61 pazienti (44%) con miopatia di Bethlem
- 21 pazienti (15%) con fenotipo intermedio/miopatia di Bethlem
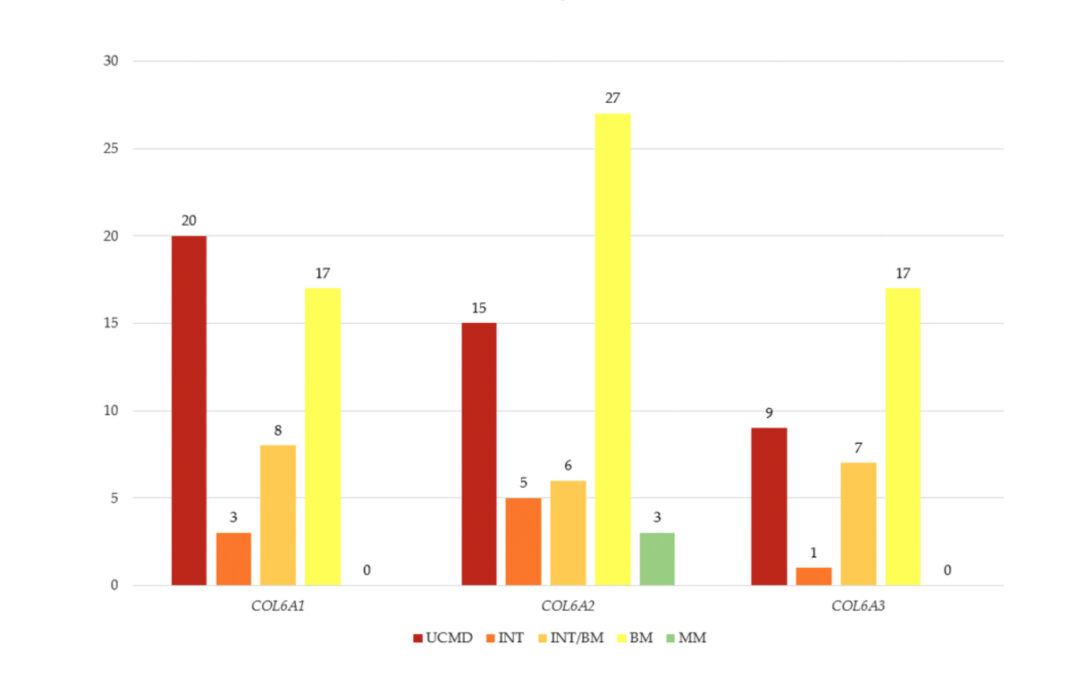
Inoltre, 3 pazienti (2%) presentavano un fenotipo di miopatia con miosclerosi (Figura 2a – Fonte: https://www.mdpi.com/2218-273X/15/10/1426).
Figura 2 – Fonte: https://www.mdpi.com/2218-273X/15/10/1426
a) Geni COL6A e fenotipi osservati nella nostra coorte.
b) Numero di varianti nei geni COL6A per ciascun fenotipo.
Le varianti del gene COL6A1 erano associate a tutti i fenotipi eccetto la miopatia con miosclerosi: 20 pazienti con distrofia muscolare congenita di Ullrich, 3 con forma intermedia, 8 con fenotipo intermedio/miopatia di Bethlem e 17 con miopatia di Bethlem.
Il fenotipo MM era presente solo nella coorte del COL6A2, dove predominavano i fenotipi meno gravi: 15 pazienti con distrofia di Ullrich, 5 con forma intermedia, 6 con fenotipo intermedio/miopatia di Bethlem, 27 con miopatia di Bethlem e 3 con miopatia con miosclerosi.
Nel gene COL6A3 si osservava una prevalenza simile di fenotipi meno gravi: 9 distrofia di Ullrich, 1 forma intermedia, 7 fenotipo intermedio/miopatia di Bethlem, 17 miopatia di Bethlem (Figura 2b).
3.3 Geni COL6A, fenotipi e modalità di trasmissione
La maggior parte dei pazienti (108 su 138, 78,3%) presentava una variante monoallelica di un gene COL6A, suggerendo una trasmissione dominante o mutazione de novo.
Varianti multiple (due o più) sono state identificate in 30 pazienti (21,7%), indicando una trasmissione recessiva.
Sebbene le varianti monoalleliche fossero prevalenti in tutti i geni COL6A, le varianti bialleliche erano significativamente rappresentate nel gene COL6A2: 24 pazienti presentavano varianti bialleliche in COL6A2, mentre solo 3 pazienti avevano varianti bialleliche in COL6A1 o COL6A3.
Le varianti bialleliche erano associate esclusivamente a distrofia muscolare congenita di Ullrich nella coorte COL6A1, mentre nelle coorti COL6A2 e COL6A3 erano legate a diversi fenotipi, con la miopatia di Bethlem predominante nella coorte COL6A2 (Figura 3).
Figura 3 – Fonte: https://www.mdpi.com/2218-273X/15/10/1426
Numero di pazienti con varianti monoalleliche o bialleliche nei geni COL6A secondo ciascun fenotipo.
3.4 Varianti COL6A e distribuzione nei domini proteici
Nei geni COL6A1 e COL6A3, le mutazioni identificate erano prevalentemente localizzate nei domini N-terminali e nei domini a tripla elica, mentre erano rare nel dominio C-terminale.
Nel gene COL6A2, le mutazioni erano distribuite lungo tutto il gene, con prevalenza nel dominio C-terminale.
La distribuzione delle mutazioni nei diversi domini dei tre geni è riportata in Figura 4 – Fonte: https://www.mdpi.com/2218-273X/15/10/1426.
In dettaglio:
- Per la catena alfa1(VI), il dominio a tripla elica era il più colpito, con varianti missenso (sostituzioni di residui di glicina) e varianti di splicing come mutazioni più comuni.
- Per la catena alfa2(VI), le varianti erano equamente distribuite tra il dominio a tripla elica e il dominio C-terminale, con prevalenza di mutazioni missenso in entrambe le regioni.
- Per la catena alfa3(VI), le varianti, principalmente missenso, erano equamente distribuite tra il dominio a tripla elica e l’ampia regione N-terminale estesa.
Figura 4 – Fonte: https://www.mdpi.com/2218-273X/15/10/1426
Rappresentazione schematica della distribuzione delle mutazioni nei diversi domini dei tre geni COL6A.
Il diagramma innovativo, chiamato Chord (Microsoft Corporation), permette di visualizzare intuitivamente le relazioni tra più variabili: in questo caso, tra i tre domini (N-terminale, tripla elica e C-terminale) dei geni COL6A1, COL6A2 e COL6A3 e i diversi tipi di varianti.
Ogni variabile, assegnata a un codice colore, è collocata in un punto preciso, definito come nodo, lungo un layout circolare e connessa ad altri nodi tramite archi, detti letteralmente “corde”. Ogni connessione ha un valore, in questo caso corrispondente al numero di varianti, rappresentato proporzionalmente dalla dimensione dell’arco: archi più spessi indicano connessioni più numerose, archi più sottili connessioni meno significative.
4. Discussione
Presentiamo il più ampio studio nazionale che descrive il panorama genetico dei pazienti non correlati con miopatie da deficit di collagene VI. La coorte di 138 pazienti proveniva da 21 centri diagnostici italiani, distribuiti su tutto il territorio nazionale, con diagnosi raccolte in un arco temporale di 15 anni (2007–2022).
L’identificazione delle mutazioni si è basata su sequenziamento tradizionale o di nuova generazione, a seconda dell’evoluzione delle tecnologie nel tempo. Solo in pochi casi è stata utilizzata una array CGH personalizzata. Questo implica che la presenza di variazioni del numero di copie nei geni COL6A non sia stata indagata completamente; tuttavia, secondo il Database delle Mutazioni Geniche Umane, le grandi riarrangiamenti genomici rappresentano solo circa il 5% dei casi di miopatia da collagene VI. Nella nostra coorte, la proporzione di varianti nei geni COL6A1 (35%), COL6A2 (40%) e COL6A3 (25%) è sostanzialmente sovrapponibile a quanto già descritto in altri paesi occidentali (38%, 44% e 18%, rispettivamente). Risultati simili sono stati osservati in una coorte egiziana di 23 pazienti, mentre in cohorti spagnole, americane e asiatiche si osservano differenze nella predominanza di alcuni geni. Questi dati non supportano l’esistenza di differenze chiare legate a popolazioni o etnie.
Il gene COL6A3 sembra essere il meno rappresentato, probabilmente perché le catene α5(VI) e α6(VI), altamente omologhe ad α3(VI), possono sostituirla nell’assemblaggio con α1(VI) e α2(VI), compensandone la perdita se mutata. Il nostro scenario mutazionale, comprendente 104 varianti diverse, conferma la notevole eterogeneità allelica delle miopatie da collagene VI. Secondo il database, oltre 2000 varianti patogenetiche sono state riportate, principalmente mutazioni puntiformi, che rappresentano circa l’82% del totale. Nella nostra coorte, lo spettro delle varianti includeva missenso, splicing, frameshift, nonsense e delezioni/inserzioni in-frame. Il gene COL6A2 è stato l’unico in cui abbiamo osservato l’intero spettro di varianti, mostrando la più alta variabilità allelica con 52 varianti diverse.
Le varianti missenso erano le più frequenti (52,9% del totale), seguite da quelle di splicing. Quasi tutte le varianti di COL6A1 e COL6A3 erano monoalleliche, mentre nel gene COL6A2 erano comuni le varianti bialleliche, associate sia a fenotipi gravi (distrofia di Ullrich) che meno gravi (miopatia di Bethlem). Tra le varianti missenso, le sostituzioni eterozigoti di glicina interrompenti la sequenza tripla Gly-X-Y sono riconosciute come patogenetiche e rappresentano il 49% delle varianti missenso; tra queste, le varianti p.(Gly284Arg) e p.(Gly293Arg) di COL6A1 ricorrono frequentemente nella coorte, associate all’intero spettro fenotipico. La variabilità fenotipica nelle miopatie da collagene VI è comune e ben documentata. Fattori genetici modificatori, epigenetici, ambientali o mosaicismi potrebbero contribuire a questa variabilità. Gruppi di pazienti con lo stesso genotipo ma severità diversa rappresentano uno strumento utile per studiare i meccanismi sottostanti.
Le varianti di splicing erano il secondo tipo più comune (25,9%), mentre in altri studi risultavano predominanti. Quattro pazienti femmine della nostra coorte portavano la variante intronica profonda c.930+189C>T nel COL6A1, nota per generare un pseudo-esone in-frame con effetto dominante, causando un fenotipo severo di distrofia di Ullrich e perdita precoce della deambulazione. Questa variante è rilevante anche per possibili terapie di salto di esone. Le varianti nonsense, frameshift, piccole delezioni o inserzioni in-frame erano meno frequenti, principalmente nel gene COL6A2, spesso associate a fenotipi recessivi e più gravi. Le miopatie da collagene VI derivano da mutazioni recessive con perdita di funzione o, più frequentemente, da varianti dominanti con effetto negativo dominante nei tre geni principali. Queste ultime comprendono inserzioni/delezioni in-frame o mutazioni puntiformi sostituenti glicina, che interrompono il motivo triplo Gly-X-Y. Le mutazioni nonsense o tronche possono anch’esse avere effetto negativo dominante se sfuggono al decadimento mediato da nonsense e si assemblano con catene normali, alterando le strutture supramolecolari.
In un paziente con miopatia di Bethlem, abbiamo identificato tramite array CGH personalizzata una delezione nell’intron 1A del COL6A2 in eterozigosi composta con una piccola delezione nell’esone 28, evidenziando l’importanza di questo strumento diagnostico complementare, soprattutto nelle forme recessive in cui solo un allele mutato è rilevato dal sequenziamento standard. Nella nostra serie, i fenotipi più lievi (miopatia di Bethlem e forma intermedia) erano prevalenti (59%), probabilmente grazie all’attività di reti cliniche di eccellenza, come la Rete Europea di riferimento per le malattie neuromuscolari, che favoriscono il riconoscimento e la corretta classificazione dei pazienti.
Nonostante non siano state identificate correlazioni genotipo-fenotipo nette, le varianti in COL6A1 tendevano ad associarsi a fenotipi più gravi, mentre quelle in COL6A2 e COL6A3 a fenotipi meno severi. La maggioranza dei pazienti presentava varianti monoalleliche, suggerendo ereditarietà dominante o de novo (78,3%), mentre il 21,7% presentava varianti bialleliche. Nella nostra coorte, le varianti bialleliche in COL6A1 determinavano invariabilmente il fenotipo severo di distrofia di Ullrich, mentre in COL6A2 e COL6A3 erano associate a tutti i fenotipi. La scoperta di casi recessivi lievi nella miopatia di Bethlem, principalmente nel gene COL6A2 e concentrati nel dominio C-terminale della catena α2(VI), suggerisce che le varianti bialleliche in COL6A2 possano essere meglio tollerate, portando a fenotipi meno gravi.
Per quanto riguarda i domini proteici coinvolti, le varianti in COL6A1 e COL6A3 erano localizzate principalmente nei domini N-terminale e tripla elica, mentre in COL6A2 erano distribuite lungo tutto il gene, con prevalenza nel dominio C-terminale. Questo è coerente con studi su larga scala, che mostrano come il dominio C-terminale sia cruciale per l’allineamento corretto del tetramero e per interazioni eterotipiche con altre molecole della matrice. Le catene di collagene VI si associano inizialmente tramite il dominio C-terminale, formando un monomero a tripla elica; i monomeri si assemblano in dimeri antiparalleli, quindi in tetrameri, che vengono secreti e organizzati in microfibrille di collagene VI. Infine, il gene COL6A2 appare il più conservato evolutivamente tra le specie, dal pesce ai primati, mentre COL6A1 e COL6A3 sono assenti in alcune specie come scimpanzé e ratti. Questo aspetto evolutivo potrebbe spiegare il panorama unico di mutazioni e associazioni fenotipiche del COL6A2.
5. Conclusioni
A nostra conoscenza, questo studio descrive la più ampia coorte di pazienti genotipizzati a livello mondiale, diagnosticati in un arco temporale di 15 anni in 21 centri diagnostici italiani. I dati raccolti confermano quanto già osservato in precedenti studi: esiste una grande variabilità del quadro clinico, un’elevata eterogeneità allelica e una conseguente complessità nella diagnosi delle miopatie da deficit di collagene VI. Recenti innovazioni nella genetica molecolare e approcci innovativi, incluso l’uso dell’intelligenza artificiale, hanno fornito un supporto significativo nel processo diagnostico di condizioni caratterizzate da forte eterogeneità, come le miopatie da collagene VI.
Molto importante è anche la strategia di networking offerta dalla Rete Europea di riferimento per le malattie neuromuscolari, che ha migliorato l’accuratezza diagnostica, la capacità di identificare i fenotipi più lievi e di categorizzare i pazienti in sottogruppi. Grazie a questa rete, i professionisti della salute possono interfacciarsi facilmente tra loro e utilizzare strumenti digitali dedicati, come la piattaforma digitale per la gestione clinica dei pazienti, utile per discutere casi clinici complessi o atipici. Un risultato importante e nuovo emerso dal nostro studio riguarda le peculiarità del gene COL6A2, che lo distinguono dai geni COL6A1 e COL6A3. In particolare:
- È il gene più frequentemente coinvolto nella nostra coorte;
- Mostra la maggiore variabilità allelica e l’intero spettro di tipi di varianti;
- Presenta varianti sia dominanti sia recessive, queste ultime più comuni rispetto agli altri due geni;
- La distribuzione delle varianti lungo la catena α2(VI) è caratteristica: non solo nel dominio N-terminale della tripla elica, ma anche frequentemente nel dominio C-terminale, cruciale per l’assemblaggio proteico.
Sebbene il numero di pazienti riportati finora sia limitato, è interessante notare che il gene COL6A2 è l’unico coinvolto nei pazienti con miopatia da miosclerosi della nostra coorte, suggerendo che dovrebbe essere il primo gene da investigare quando il fenotipo è altamente indicativo. Questa diversità è sorprendente, considerando che COL6A1 e COL6A2 condividono una sequenza e una struttura proteica molto simili, si trovano entrambe nella regione cromosomica 21q22.3 e derivano da una duplicazione genica ancestrale. Tuttavia, esistono differenze evolutive e strutturali: il gene COL6A2 è più antico nella filogenesi animale, è l’unico COL6A preservato in tutti i vertebrati senza perdite geniche e mostra una diversa organizzazione introne-esone nei domini globulari rispetto a COL6A1, pur conservando la struttura degli esoni della tripla elica. Queste differenze potrebbero riflettere funzioni diverse dei due geni e meritano ulteriori indagini per comprendere la loro correlazione con il pattern di mutazioni del COL6A2 e le implicazioni nella fisiopatologia delle miopatie da collagene VI.
Autori – Fernanda Fortunato, Laura Fiocco, Alice Margutti, Marcella Neri, Adele D’Amico, Enrico Bertini, Enzo Ricci, Eugenio Maria Mercuri, Marika Pane, Roberto Massa, Giulia Greco, Angela Alessandra Ferlini, Francesca Gualandi, Cristina Cereda, Antonella Pini, Luciano Merlini, et al.